2022 VINNOVA | 第七届血管创新论坛
4月22日,第七届血管创新论坛(VINNOVA 2022)在线上隆重召开,大会由中国研究型医院学会血管医学专委会、中国医疗器械行业协会血管器械分会、北京博瑞血管健康公益基金会共同主办,数百名行业专家参与发言。
会议为临床专家、科研院所、政策制定者、产业界搭建了交流平台,共同探讨血管疾病前沿技术和发展动态,为先行者提供探索的方向,通过业内跨界交流推动我国血管医学领域的快速发展。
如何在合规框架下加速创新产品临床试验
奥泰康扎根创新医疗器械和高端医疗器械,提供临床注册全程解决方案。创新论坛中,中国医学科学院阜外医院-李卫教授的发言也为创新产品的临床试验设计提供了解决方案,在此,我们对李卫教授的讲解进行了拓展解析。

李卫
中国医学科学院阜外医院
中国医学科学院北京协和医学院阜外医院研究员、博士生导师,国家心血管病中心医学统计部主任,法国生物统计学博士,香港中文大学公卫学院客座教授,中国医疗器械行业协会医学数据分析专业委员会主任委员,中华医学会心血管病分会信息化学组副组长,中国医药教育协会医药统计专业委员会常委,国家药品监督管理局药物及医疗器械审评专家,国家科技部、国家卫健委临床研究审评专家,中国临床试验数据管理学组及统计学组成员,国际临床试验生物统计学组成员,主编或主译临床试验统计方法学论著3部,在临床研究设计及评价方面具有丰富经验。
如何在合规框架下加速创新产品临床试验-视频

PPT Page 1

PPT Page 2
2017年10月8日,为鼓励药品医疗器械科技创新,提高上市产品质量和产业竞争力,满足公众临床需要,中共中央办公厅、国务院办公厅印发了《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》。在多方面深化了审批制度,包括改革临床试验管理、加快上市审评审批、促进药品创新和仿制药发展、加强药品医疗器械全生命周期管理、提升技术支撑能力、加强组织实施等。
政策链接:关于深化审评审批制度改革鼓励药品医疗器械创新的意见.doc
PPT Page 3
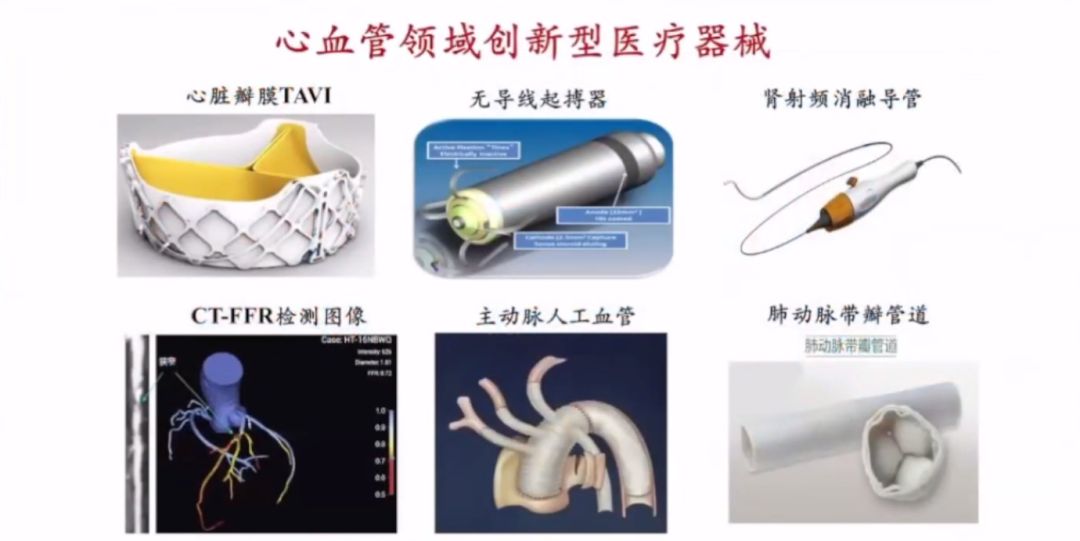
自政策实施以来,我国心血管领域有多款医疗器械通过创新审批,为临床带来新选择,包括心脏瓣膜TAVI、CT-FFR等创新产品。

PPT Page 4
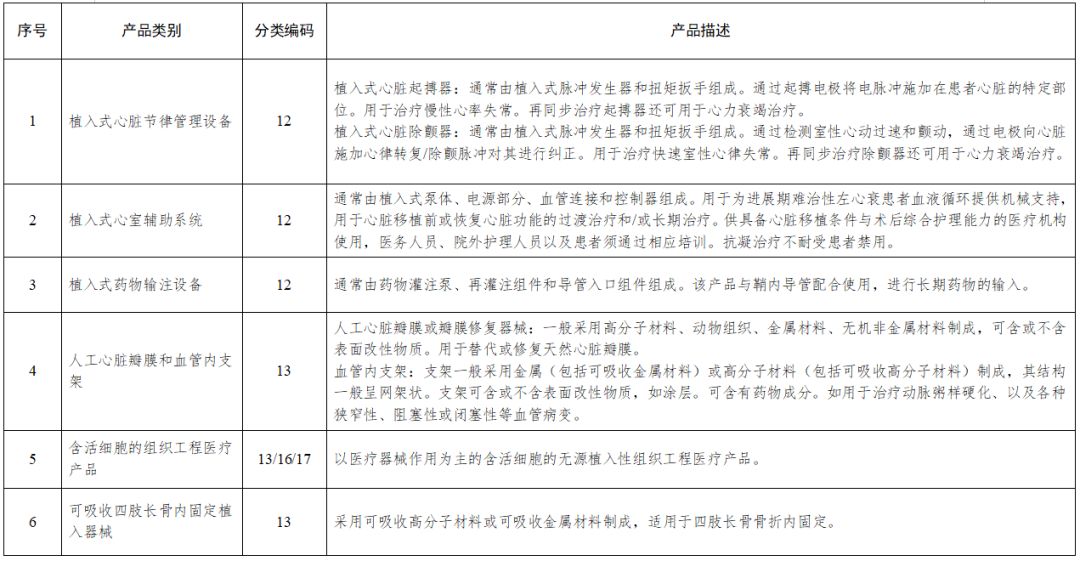
根据我国医疗器械分类,二类、三类要需要做临床试验,对人体具有较高风险的第三类医疗器械进行临床试验,应当经国务院药品监督管理部门批准,拿到高风险医疗器械临床试验审批证明。列入《需进行临床试验审批的第三类医疗器械目录》中的医疗器械应当在中国境内进行临床试验,包括:
●植入式心脏节律管理设备
●植入式心室辅助系统
●植入式药物输注设备
●人工心脏瓣膜和血管内支架、
●含活细胞的组织工程医疗产品
●可吸收四肢长骨内固定植入器械
政策链接:需进行临床试验审批的第三类医疗器械目录(2020年修订版).doc
《需进行临床试验审批的第三类医疗器械目录》

PPT Page 5
2014年2月7日,为保障医疗器械的安全有效,鼓励医疗器械的研究与创新,促进医疗器械新技术的推广应用,推动医疗器械产业发展,国家药监局制定了《创新医疗器械特别审批程序(试行)》,政策从创新产品定义、提交资料、证明性文件、等方面规定了创新器械审批流程和要点。

PPT Page 6
通过创新审批后,可享有审评部门早期介入、专人负责、提供指导、优先审评、加强沟通等诸多VIP待遇,为企业节约大量时间,缩短产品上市时间。

PPT Page 7
李卫教授讲到,虽然是创新医疗器械,国家药监局规定也要按照医疗器械临床试验相关规定的要求去执行,评价产品的安全性和有效性,创新政策的推出是为了加速审评过程,但不会降低审批标准。

PPT Page 8
2018年1月8日,国家食品药品监督管理总局为贯彻落实中共中央办公厅、国务院办公厅《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号),加强医疗器械产品注册工作的管理,进一步提高注册审查质量,鼓励医疗器械研发创新,制定了医疗器械临床试验设计指导原则。原则中阐述了临床试验设计的基本类型及特点、受试对象、评价指标、比较类型和检验假设、样本量估算等,为方案设计者提供依据。
政策链接:医疗器械临床试验设计指导原则.doc
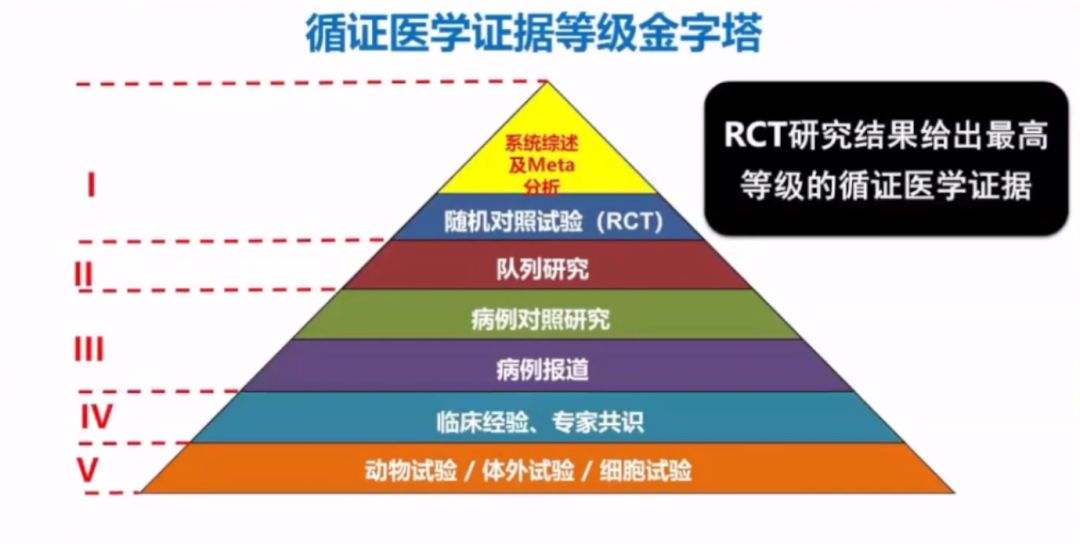
PPT Page 9
李卫教授表示,根据循证医学证据级别金字塔中最高等级的I级证据显示,基于良好设计的随机对照试验(randomized controlled trial,RCT)最能够评价器械的安全性和有效性。

PPT Page 10
李卫教授总结到,以上市注册为目的的创新型医疗器械临床试验需在符合伦理和临床可行的情况下,应进行最高循证等级的前瞻性、随机对照研究。

PPT Page 11
李卫教授讲到,美国FDA针对全新的心血管长期植入器械,也要做前瞻性、随机对照临床试验。创新产品无同类产品可对照,可与常规的药物/手术治疗比较的优效性设计,也就是最小负担原则。
拓展学习:
●定义:
最小负担原则,即“用最少量的必要信息,在适当的时间,以最有效的方式,恰当地解决相关监管问题或事项”。最小负担原则适用于所有医疗器械及其全生命周期。
●主要内容:
①审评机构要求提供必要的最少信息,以充分解决当前的审评问题;
②企业在提交注册申报资料时,应保证审评机构负担最小;
③采用最有效的方法解决问题,如充分的沟通交流,或者制定补正通知书的指导原则,提出补正通知书的要素“证据、差距、原因、期望”,清晰、简明地传达审评要求、期望、过程、政策和决定以及背后的基本原理;
④在合适的时机提出合适的信息要求,如在合适和可行的情况下利用真实世界数据减少对临床数据的要求;
⑤与技术相适应并充分考虑特殊的产品创新周期、证据需求及患者需求;
⑥在合理和可行的前提下利用和参考其他国家和地区的数据和监管部门决策;
⑦在实现国际医疗器械监管趋同和协调统一中应用最小负担原则,如认可和使用国际和其他标准组织发布的标准。
●优点
应用最小负担原则有利于减轻审评负担,将有限的审评审批资源聚焦高风险产品,通过最有效的方式在最恰当的时机,利用最少量的必要信息进行有效监管,更好地促进最新的高质量、安全、有效和经济的医疗器械快速应用于临床。

PPT Page 12
很多情况下,若无同类产品和治疗手段可做对照,单组目标值的试验方法不失为一种替代、妥协性的研究设计方法。
拓展学习:
随机对照临床试验( randomized controlled trial, RCT)是临床研究的金标准,药物/医疗器械临床研究中,尤其是提供关键证据的临床研究中一般均采用
RCT。在极少数的医疗器械临床试验中,如采用随机对照试验,会存在伦理学风险,致使临床操作不可行。在此 情 况 下,单 组 目 标 值 ( single-arm objective per- formance criteria ,OPC) 临床试验不失为一种替代策略,为产品注册提供关键证据。
文献链接:单组目标值临床试验的统计学考虑.pd

PPT Page 13
在法规框架下进行创新产品的临床试验设计,李教授认为最关键的是适应症的选择问题(有无对照组),可设计出2个方案,以便和国家药监局相关部门沟通。
●随机对照(RCT):与常规治疗手段比较。